Quinoxaline amino derivatives as potential EGFR-targeted therapeutics in breast cancer: computational exploration
DOI:
https://doi.org/10.69857/joapr.v14i3.2008Keywords:
EGFR, ADMET, TNBC, MMGBSA, Breast cancer, Quinoxaline amino derivativesAbstract
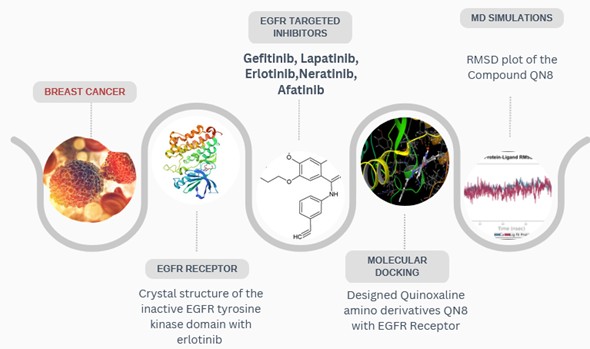
Background: Despite resistance to current tyrosine kinase inhibitors, which makes the epidermal growth factor receptor (EGFR) a recognized therapeutic target in breast cancer, there is a need for novel inhibitors. Quinoxaline compounds are a promising scaffold for next-generation EGFR inhibitors and exhibit favorable pharmacological properties. Methodology: Ten new amino quinoxaline derivatives (QN1–QN10) were systematically developed and assessed by a comprehensive in silico approach. Molecular docking was conducted on the EGFR tyrosine kinase domain (PDB ID: 4HJO) utilizing Glide XP, with erlotinib serving as the reference ligand. Drug-likeness, oral bioavailability, and synthetic accessibility were evaluated using SwissADME, whereas pkCSM predicted ADMET characteristics. The most effective candidate was subsequently corroborated by 100 ns molecular dynamics (MD) simulations utilizing GROMACS 2021.1, succeeded by MM-GBSA binding free energy assessments. Results and Discussion: According to docking data, QN8 proved the most promising inhibitor. It showed stable hydrogen bonding with key EGFR hinge residues (MET769 and ASP831) and a high binding affinity (−9.305 kcal/mol), comparable to erlotinib (−9.501 kcal/mol). Consistent RMSD and RMSF profiles from MD simulations corroborated the structural stability of the QN8–EGFR complex. According to MM-GBSA analysis, the van der Waals, lipophilic, and electrostatic contributions were the main drivers of the favorable binding free energy (-73.63 kcal/mol). Pharmacokinetic predictions showed adequate ADMET properties and good oral absorption. Conclusion: This exhaustive computational analysis highlights amino quinoxaline derivatives as promising leads for developing breast cancer drugs, identifying QN8 as a strong EGFR inhibitor with stable binding dynamics and favorable drug-like properties.
Downloads

References
Jackson K, Nkoana A, Garland K, More K, et al. Synthesis and in vitro exploration of the 8-carbo substituted 5-methoxyflavones as anti-breast and anti-lung cancer agents targeting protein kinases (VEGFR-2 & EGFR). Bioorg Chem, 153, 107875 (2024) https://doi.org/10.1016/j.bioorg.2024.107875
Lehmann BD, Jovanović B, Chen X, et al. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One, 11(6), e0157368 (2016) https://doi.org/10.1371/journal.pone.0157368
Samantaray A, Pradhan D. Novel combination therapy of Osimertinib and Tupichinol E in triple-negative breast cancer: targeting EGFR and CDK4/6 pathways. Aspects Mol Med, 5, 100069 (2025) https://doi.org/10.1016/j.amolm.2025.100069
Herdiansyah MA, Ansori ANM, Kharisma VD, Maksimiuk MR, et al. In silico study of cladosporol and its acyl derivatives as anti-breast cancer agents against estrogen receptor-α. Biosaintifika, 16, 142–154 (2024) https://doi.org/10.15294/biosaintifika.v15i1.949
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function. J Comput Chem, 31(2), 455–461 (2010) https://doi.org/10.1002/jcc.21334
Muhammed MT, Aki-Yalcin E. Molecular docking: principles, advances, and its applications in drug discovery. Lett Drug Des Discov, 21(3), 480–495 (2024) https://doi.org/10.2174/1570180819666220922103109
Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol, 9, 646–652 (2002) https://doi.org/10.1038/nsb0902-646
Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron, 99, 1129–1143 (2018) https://doi.org/10.1016/j.neuron.2018.08.011
Ejeh S, Otaru HA, Ejeh JE, John J, Ajala A, Omowanle J, et al. Computational techniques for analyzing quinoxaline-containing molecules as anti-schistosomal agents using docking, pharmacokinetics, drug-likeness, QSAR and molecular dynamics evaluation. In Silico Res Biomed, 1, 100024 (2025) https://doi.org/10.1016/j.insi.2025.100024
Badithapuram V, Nukala SK, Thirukovela NS, Dasari G, Manchal R, Bandari S. Design, synthesis, and molecular docking studies of quinoxaline derivatives as EGFR targeting agents. Russ J Bioorg Chem, 48(3), 565–575 (2022) https://doi.org/10.1134/S1068162022030220
Salem MG, Abu El-Atab SA, Elsayed EH, Mali SN, Alshwyehde HA, Almaimani G, et al. Novel 2-substituted quinoxaline analogs with antiproliferative activity against breast cancer. RSC Adv, 13, 33080–33095 (2023) https://doi.org/10.1039/D3RA06189B
Saritha K, Alivelu M, Mohammad M. Drug-likeness analysis and in silico ADMET profiling of compounds in Kedrostis foetidissima. In Silico Pharmacol, 12(2), 67 (2024) https://doi.org/10.1007/s40203-024-00240-1
Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem, 58(9), 4066–4072 (2015) https://doi.org/10.1021/acs.jmedchem.5b00104
Kato K, Nakayoshi T, Kurimoto E, Oda A. Molecular dynamics simulations for protein–ligand complexes obtained by docking studies. Chem Phys Lett, 781, 139022 (2021) https://doi.org/10.1016/j.cplett.2021.139022
Pentu N, Azhakesan A, Kumar PK. Insilico molecular docking and ADME/T studies of flavonol compounds against selected proteins involved in inflammation mechanism. J. Appl. Pharm. Res., 13, 95–111 (2025) https://doi.org/10.69857/joapr.v13i1.706.
Genheden S, Ryde U. MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov, 10(5), 449–461 (2015) https://doi.org/10.1517/17460441.2015.1032936
Park JH, Liu Y, Lemmon MA, Radhakrishnan R. Erlotinib binds inactive and active conformations of EGFR tyrosine kinase domain. Biochem J, 448(3), 417–423 (2012) https://doi.org/10.1042/BJ20121513
Wanode DM, Bhendarkar KP, Khedekar PB. Discovery of pyrazole–pyrazoline derivatives as VEGFR-2 kinase inhibitors: an in silico approach. J Appl Pharm Sci, 16(2), 122–135 (2025) https://doi.org/10.7324/JAPS.2026.256031
Sharma VK, Nandekar PP, Sangamwar A, Pérez-Sánchez H, Agarwal SM. Structure-guided design and binding analysis of EGFR inhibiting analogues. RSC Adv, 6, 105920–105939 (2016) https://doi.org/10.1039/C6RA20811C
Mousavinejad SN. Molecular docking comparison of icotinib and erlotinib as EGFR inhibitors. Zahedan J Res Med Sci, 28(1), e165680 (2026) https://doi.org/10.5812/zjrms-165680
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics and drug-likeness of small molecules. Sci Rep, 7, 42717 (2017) https://doi.org/10.1038/srep42717
Bakchi B, Krishna AD, Sreecharan E, Ganesh VBJ, Niharika M, Maharshi S, et al. Applications of SwissADME web tool in medicinal chemistry. J Mol Struct, 1259, 132712 (2022) https://doi.org/10.1016/j.molstruc.2022.132712
Xu ZY, Li JL. Drug–drug interactions with EGFR tyrosine kinase inhibitors in NSCLC treatment. Onco Targets Ther, 12, 5467–5484 (2019) https://doi.org/10.2147/OTT.S194870
Kucharczuk CR, Ganetsky A, Vozniak JM. Drug–drug interactions, safety, and pharmacokinetics of EGFR tyrosine kinase inhibitors. J Adv Pract Oncol, 9(2), 189–200 (2018) https://doi.org/10.6004/jadpro.2018.9.2.5
Shetty SR, Debnath S, Majumdar K, Rajagopalan M, Ramaswamy A, Das A. Virtual screening, molecular dynamics simulations, and in vitro validation of EGFR inhibitors as breast cancer therapeutics. Bioorg Chem, 153, 107849 (2024) https://doi.org/10.1016/j.bioorg.2024.107849
Published
How to Cite
Issue
Section
Copyright (c) 2026 Abitha H, D. Kumudha, Bhuvaneswari Sivaraman, M. K. Kathiravan

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.














